We are thrilled to announce the support for running Snakemake workflows on REANA reproducible analysis platform, starting from the REANA 0.8.0 release. Snakemake joins CWL and Yadage as another complete workflow definition language that REANA users can use to run their analysis workflows.
What is Snakemake?
Snakemake is a workflow engine that originated in bioinformatics in 2012. Snakemake workflows are defined using Snakefile, similar in structure to Makefile. The Snakefile uses a domain-specific language (DSL) with a syntax similar to YAML and Python. Snakemake workflows are described in terms of rules. Each rule specifies the list of inputs, which shell or Python commands to execute, the conditions such as container to use, the number of threads, as well as the list of output files of the rule. Snakemake creates a Directed Acyclic Graph (DAG) representing the data analysis workflow for the desired target rule and plans the job execution for the rules. The REANA platform now includes support for Snakemake workflow definitions and executes the workflow and its jobs using the regular REANA compute backends.
How to create and run Snakemake workflows in REANA
Let us take the ROOT6 RooFit demo analysis as example of how you can create and run Snakemake workflow.
Create Snakefile
The demo analysis workflow consists of two steps where we generate the
data (rule gendata) and then fit it against a theoretical model
(rule stepdata).
The workflow contains several input parameters such as the number of
events to generate or the name of the data-generating and fitting
scripts. In order to parametrise the Snakemake workflow, we create a
new inputs.yaml file containing key-value pairs as follows:
# inputs.yaml
events: 20000
fitdata: code/fitdata.C
gendata: code/gendata.C
The workflow produces output plots which we shall specify in the
overarching rule all.
The resulting Snakefile can then be constructed as follows:
# Snakefile
rule all:
input:
"results/data.root",
"results/plot.png"
rule gendata:
input:
gendata_tool=config["gendata"]
output:
"results/data.root"
params:
events=config["events"]
container:
"docker://docker.io/reanahub/reana-env-root6:6.18.04"
shell:
"mkdir -p results && root -b -q '{input.gendata_tool}({params.events},\"{output}\")'"
rule fitdata:
input:
fitdata_tool=config["fitdata"],
data="results/data.root"
output:
"results/plot.png"
container:
"docker://docker.io/reanahub/reana-env-root6:6.18.04"
shell:
"root -b -q '{input.fitdata_tool}(\"{input.data}\",\"{output}\")'"
Create reana-snakemake.yaml
We need to create the usual REANA specification file
reana-snakemake.yaml describing the overall analysis structure.
In order to indicate that the workflow we are writing is a Snakemake
workflow, the specification file must set the workflow.type
directive to snakemake:
# reana-snakemake.yaml
...
workflow:
type: snakemake
...
The workflow parameters created above in the new inputs.yaml file
are set in the inputs.parameters.input directive:
# reana-snakemake.yaml
...
inputs:
...
parameters:
input: workflow/snakemake/inputs.yaml
...
Finally, we indicate the path to Snakefile in the workflow.file
directive:
# reana-snakemake.yaml
...
workflow:
...
file: workflow/snakemake/Snakefile
...
The resulting complete reana-snakemake.yaml looks as follows:
# reana-snakemake.yaml
version: 0.8.0
inputs:
files:
- code/gendata.C
- code/fitdata.C
directories:
- workflow/snakemake
parameters:
input: workflow/snakemake/inputs.yaml
workflow:
type: snakemake
file: workflow/snakemake/Snakefile
outputs:
files:
- results/plot.png
The important Snakemake-specific parts are highlighted. The rest of the specification looks as usual, defining input files and directories and the output plot.
Run the workflow
Prior to running our workflow let us validate the created
specification by means of using the reana-client validate --environments command. The extra validation option will validate
also workflow parameters and
environments.
$ reana-client validate -f reana-snakemake.yaml --environments --pull
==> Verifying REANA specification file... ~/src/reanahub/reana-demo-root6-roofit/reana-snakemake.yaml
-> SUCCESS: Valid REANA specification file.
Job stats:
job count min threads max threads
------- ------- ------------- -------------
all 1 1 1
fitdata 1 1 1
gendata 1 1 1
total 3 1 1
==> Verifying REANA specification parameters...
-> SUCCESS: REANA specification parameters appear valid.
==> Verifying workflow parameters and commands...
-> SUCCESS: Workflow parameters and commands appear valid.
==> Verifying dangerous workflow operations...
-> SUCCESS: Workflow operations appear valid.
==> Verifying environments in REANA specification file...
-> SUCCESS: Environment image reanahub/reana-env-root6:6.18.04 has the correct format.
-> SUCCESS: Environment image reanahub/reana-env-root6:6.18.04 exists locally.
-> SUCCESS: Environment image reanahub/reana-env-root6:6.18.04 exists in Docker Hub.
-> INFO: Environment image uses UID 0 but will run as UID 1000.
Everything looks good, so let us run the example, check workflow status and the output plots:
$ reana-client run -w roofit-snakemake -f reana-snakemake.yaml --skip-validation
==> Creating a workflow...
roofit-snakemake.1
==> Uploading files...
==> SUCCESS: File /code/gendata.C was successfully uploaded.
==> Starting workflow...
==> SUCCESS: roofit-snakemake.1 has been queued
...
$ reana-client status -w roofit-snakemake
NAME RUN_NUMBER CREATED STARTED ENDED STATUS PROGRESS
roofit-snakemake 1 2021-10-04T14:00:56 2021-10-04T14:01:08 2021-10-04T14:01:20 finished 2/2
$ reana-client ls -w roofit-snakemake --filter name=results
NAME SIZE LAST-MODIFIED
results/data.root 154455 2021-10-04T14:01:09
results/plot.png 15450 2021-10-04T14:01:16
We can visualise the plot:
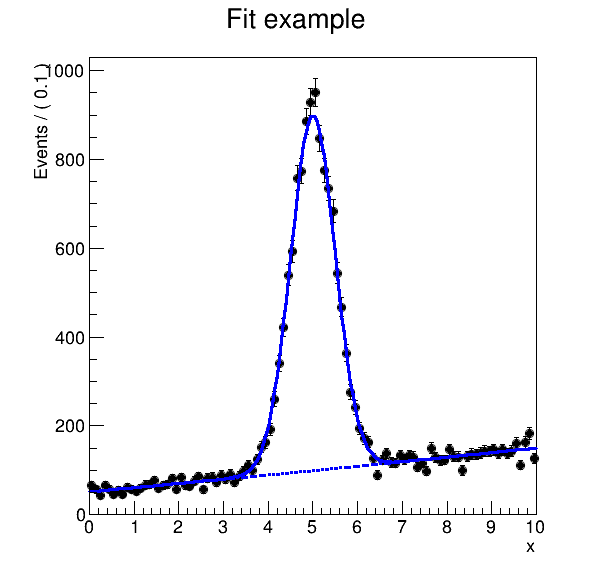
Inspect workflow execution report
REANA generates a Snakemake report when a workflow succeeds, so that you can have a more detailed view of the workflow computational graph, the overview of different steps, their run times, etc.
The generated report can be found under the file name
report.html. You can customise the report name via the report
operational
option.
Let us download the generated report and visualise it in our favourite browser:
$ reana-client download -w roofit-snakemake 'report.html'
==> SUCCESS: File report.html downloaded to ~/src/reanahub/reana-demo-root6-roofit.
$ firefox report.html

We can click on each node to find more information about each step such as inputs, outputs, environment, commands, etc.
Snakemke in the REANA ecosystem
The integration of Snakemake in the REANA platform allows Snakemake
users to profit from the other regular REANA features in their
workflows. For example, it is possible to execute hybrid workflows by
simply setting appropriate compute_backend parameter in the
resources directive of the given Snakemake rule.
As an example, let us amend the above Snakefile to instruct REANA to
run the data generation step on HTCondor compute backend instead of on
Kubernetes. It is sufficient to alter few line in the above rule:
# Snakefile-htcondor
rule gendata:
input:
gendata_tool=config["gendata"]
output:
"results/data.root"
params:
events=config["events"]
container:
"docker://docker.io/reanahub/reana-env-root6:6.18.04"
resources:
compute_backend="htcondorcern",
htcondor_max_runtime="espresso"
shell:
"mkdir -p results && root -b -q '{input.gendata_tool}({params.events},\"{output}\")'"
In this way, we can simply create hybrid Snakemake workflows where some parts of calculations are executed on HTCondor high-throughput compute backend, other parts on Slurm high-performance compute backend, and yet other parts on the default Kubernetes compute backend.
Please try out the new Snakemake support in REANA and please do not hesitate to report any issues you may encounter using our GitHub issue tracker.
See also: